
All data from these pages are results of my thesis work (download the manuscript - 9Mo, written in French).
This work is a numerical study of point defect diffusion in semi-conductors such as Si and SiGe. As macroscopic gradients of concentrations are the result of atomic moves, a multi-scale approach can be used.
Ab initio calculations are highly useful when investigating atomic interactions, and, when linked with geometric minimization algorithms they give access to stable and transition states. The macroscopic movement can then be simulated using kinetic Monte Carlo calculations.
We detail, in this work, the geometry and the energetic cost of most stable points defects of Si and SiGe. This includes vacancies, dumbbell [110] interstitials, hexagonal interstitals and four-folded coordinated defects. We study their movements and use this information in thermodynamical simulations to show that several regimes exist for the diffusion, depending on the interactions between mediators. In the case of the vacancy assisted diffusion, the differences observed in diffusivity are explained by the existence of vacancy clusters and their dissociation mechanisms.
This study shows that the coupling between atomistic and macroscopic simulations is required to explain diffusion mechanisms.

Fig. 1: formation energies of vacancy clusters in silicon, calculated by ab initio methods (DFT-LDA, 216 sites computed at Γ point. (Enlarge the figure)
Using DFT-LDA calculations, we investigate the effect of vacancy clustering in silicon. This confirms the previous results from tigh-binding calculations that vacancy clusters are favored ; and that some clusters are even more stable than others (emphasing magic numbers such as 2 or 6), see figure 1.
Our results are compared with those of Bongiorno et al. [Europhysics Letters 43, 695 (1998)] and of Staab et al. [Physical Review B 65, 115210 (2002)].
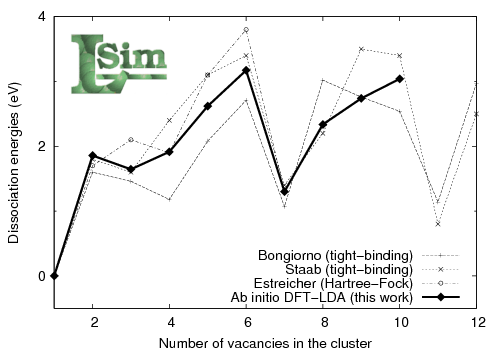
Fig. 2: dissociation energies for vacancy clusters in silicon. (Enlarge the figure)
Comparing dissociation energies, tight-binding and ab initio are still in good agreement discussing which cluster is most stable but they can be different up to 0,7eV when numbers are involved (see figure 2). This discrepancy is quite important when diffusion is studied and ab initio results should be used instead when small clusters are involved.

Fig. 3: Plot of square distance versus simulated time showing two mechanisms. (Enlarge the figure)
We studied vacancy diffusion using a Kinetic Monte Carlo on Lattice method. The energies used in the KMC are taken from configurations calculated with ab initio. These simulations highlight an important reduction of the diffusivity coefficient due to clusters and different competitive mechanisms (see figure 3).
These simulations have been done with a KMC code I developped for this specific problem. Different sizes of clusters have been tested and clusters with 4 or 5 vacancies required to implement a special algorithm to avoid movements within basins of energy. Indeed such movements would have stop the diffusion by only performing localized changes at high hoping rates.
With ab initio calculations and NEB (Nudged-Elastic Band) method, we investigated and quantified all possible movements linking most stable defects in SiGe. This result in the three classes represented in figure 4-{a, b, c}. For each kind of movement, we carefully computed the barrier for when one silicon atom and one germanium atom are involved.

Fig. 4-a: Dumbbell to hexagonal movements.

Fig. 4-c: Hexagonal to hexagonal movements.

Fig. 4-b: Bassin movements.
damien D caliste A cea D fr) |